Hi guys! Finally, I am back with the third part of my Classical Molecular Dynamics (CMD) series. Today we will see how to solve a basic molecular dynamics equation. For those who missed my initial posts related to CMD, please see it in links below:
Part 1 (The Fundamentals)
Part 2 (The Force Field!)
And for today:
Adapted from this wiki Image.
By the way, I just found a nice short video on introduction to MD on youtube. You may want to watch it.
SUMMARY OF FIRST TWO PARTS:
CMD simulations are techniques used to mimic the motions of molecules assuming they obey Newton's laws. Since the force assumed is of the conservative nature(please see Part 1 (The Fundamentals)) we can write the force as below:
.png)
Where 'U' is the total potential energy. But you need to know the functional form of the potential energy. In the Part 2 (The Force Field!), we derived this term which is also called Force-Field. A typical force-field equation looks like below:

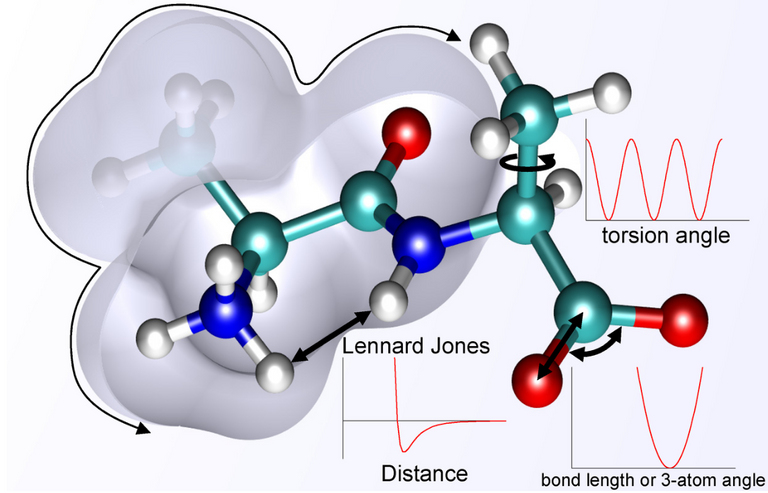
The individual components of force-field is shown in above image (adapted from here).
ENOUGH SUMMARY! LET US BEGIN.
So what do we mean by solving the MD equation?
Solving an MD equation will give you the future position coordinates and velocities of particles based on the initial coordinates and velocities(initial conditions) of the system.
The equation is as below:
We have forcefield 'U' with us now. Assume that we have all the parameters too (say from experiments). This is actually a differential equation. We need to solve this to see the evolution of motion of particles in our system.
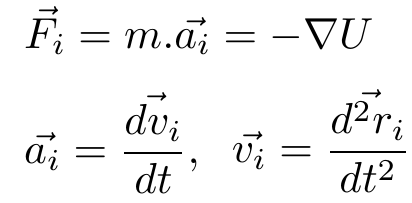
Acceleration of the particle( or atom) 'i' : ai (rate of change of velocity)
Velocity of the particle 'i' : vi (rate of change of displacement)
Force on the particle 'i' : Fi
Position on the particle 'i' : ri
Potential - 'U'
Arrows on symbols represent that they are vectors (has direction and magnitude.)
THE INTEGRATOR
So in order to solve the above equations of motion in a computer, we need to approximate them (make them discrete). One thing to be careful here is the:
- The Newton's equation of motion is reversible in time, so our integrator must reproduce this feature.
- The integrator must be symplectic in nature (it naively means there are some quantities like energy, phase space volume representing the microstates etc which has to be conserved. Will come to this point later.)
- Must be fast and reasonably has a lesser error.
We will focus on two such integrators:
- Verlet integrator
- Velocity-Verlet integrator
The figure below shows the approximation of second derivative of position a.k.a acceleration and also a visualization of a time axis.
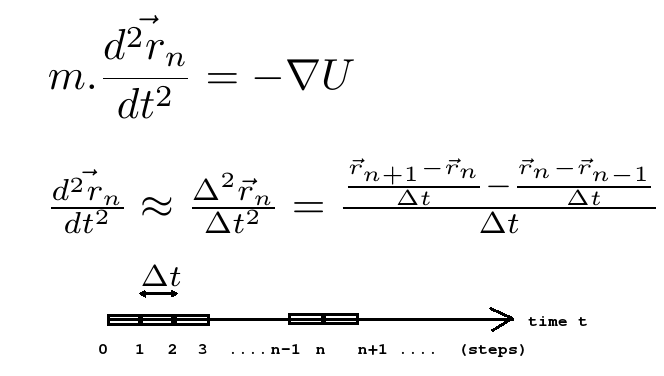
VERLET INTEGRATOR
Using Taylor's expansion we can approximate the n+1th and n-1th step of positions as below:
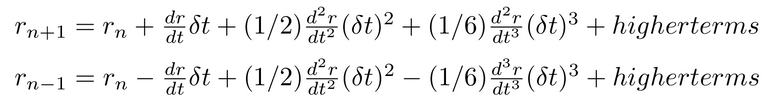
Now if you add the both of the above equations you get something as below :
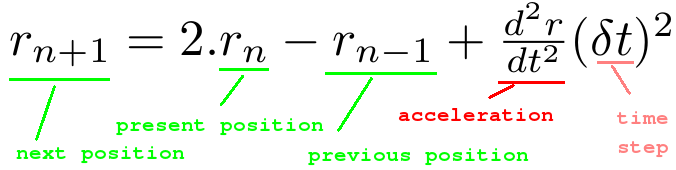
There will be an error term of order 4, which can be neglected for now. Verlet algorithm requires 2 previous position coordinates and it calculates velocity explicitly using central difference method. This is a very simple integrator which obeys symplectic and time reversible properties.
VELOCITY-VERLET INTEGRATOR
This integrator modifies Verlet algorithm to get a more accurate implementation. It starts with the n+1th position term expansion based on Taylor series and velocity term approximation in terms of accelration. Like below:
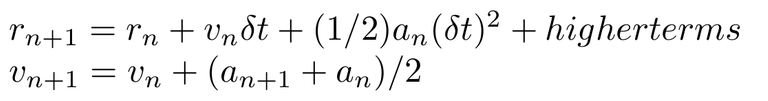
Now I will describe the iteration scheme (Algorithm):
Calculate the next middle step velocity v(n + 1/2).
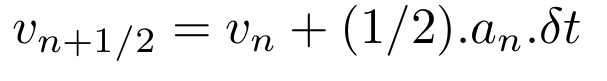
Calculate next positions r(n+1).
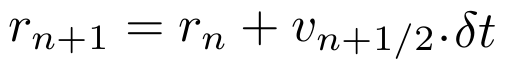
Calculate accelerations of next step from the potential 'U'.
Update the velocities to get next velocity value.
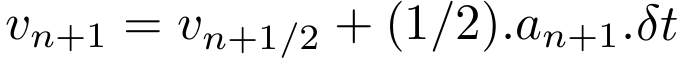
Go to 1. and repeat for next steps until the desired total time.
Choosing time step 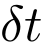
Choosing a small time step will give you a "very" accurate description, but at the expense of slow computation. So it is a trade-off between accuracy and computational efficiency. Also if the time step is too high the integration itself will spit out erroneous results. As a general rule, the time-step is kept one order less than the highest vibrational content in the system. For example, the typical frequency of molecular vibrations is in the range of 1014 Hertz. That means the time periods are in the order of 10-14 seconds. So sually the time step for all-atom classical molecular dynamics simulations are in the range of 2 fs or 1fs. (1fs = 10-15 seconds).
Hope you guys learned something new and interesting. These concepts will get start getting clear once we try out the simulation part. So wait for the experimentation section. Lots of fun awaits us in this series. If I am to summarise today's long article in one statement it is this:
Today we learned some basic techniques to predict the future positions and velocities of atoms in a molecular dynamics system, provided we know the initial coordinates and velocities.
Thats it! See you soon! Bye
Textbook references:
- "Statistical Mechanics: Theory and Molecular Simulations" by Mark E. Tuckerman
- "Molecular Modelling: Principles and Applications" by Andrew R. Leach
- "Computer Simulation of Liquids" by D. J. Tildesley and M.P. Allen [Classic! and its little bit hard for beginners]
Hi,
@dexterdev here
Please Upvote and resteem my posts if you like my content.
I would like to hear your feedback. So please do comment those.
In case you have questions, please shoot those in comments.
All images without image sources are my creations :)

Thankyou @rocking-dave for sharing the above #steemSTEM gif to community.
This is a test comment, notify @kryzsec on discord if there are any errors please.
Being A SteemStem Member
This knowledge so difficult, i thinks..
Please do ask me doubts. I can try clarifying. :)
very informative. thanks for sharing.
Thanks :) Watch out for more.
Just spotted a typo. delta-t was missing in the below equation. Thanks to rajam for spotting it:
must be